

Niemann-Pick disease
NPD; Sphingomyelinase deficiency; Lipid storage disorder - Niemann-Pick disease; Lysosomal storage disease - Niemann-Pick
Niemann-Pick disease (NPD) is a group of diseases passed down through families (inherited) in which fatty substances called lipids collect in the cells of the spleen, liver, and brain.
There are three common forms of the disease:
- Type A
- Type B
- Type C
Each type involves different organs. It may or may not involve the nervous system and breathing. Each one can cause different symptoms and may occur at different times throughout life.
Causes
NPD types A and B occur when cells in the body do not have an enzyme called acid sphingomyelinase (ASM). This substance helps break down (metabolize) a fatty substance called sphingomyelin, which is found in every cell in the body.
If ASM is missing or does not work properly, sphingomyelin builds up inside cells. This kills your cells and makes it hard for organs to work properly.
Type A occurs in all races and ethnicities. It is more common in the Ashkenazi (Eastern European) Jewish population.
Type C occurs when the body cannot properly break down cholesterol and other fats (lipids). This leads to too much cholesterol in the liver and spleen and too much of other lipids in the brain. Type C is most common among Puerto Ricans of Spanish descent.
Type C1 is a variant of type C. It involves a defect that interferes with how cholesterol moves between brain cells. This type has only been seen in French Canadian people in Yarmouth County, Nova Scotia.
Symptoms
Symptoms vary. Other health conditions may cause similar symptoms. The early stages of the disease may only cause a few symptoms. A person may never have all the symptoms.
Type A usually begins in the first few months of life. Symptoms may include:
- Abdominal (belly area) swelling by 3 to 6 months of age
- Cherry red spot at the back of the eye (on the retina)
- Feeding difficulties
- Loss of early motor skills (gets worse over time)

Type B symptoms are usually milder. They occur in late childhood or the teenage years. Abdominal swelling may occur in young children. There is almost no brain and nervous system involvement, such as loss of motor skills. Some children may have repeated respiratory infections.
Types C and C1 usually affects school-age children. However, it may occur any time between early infancy to adulthood. Symptoms may include:
- Difficulty moving limbs that may lead to unsteady gait, clumsiness, walking problems
- Enlarged spleen
- Enlarged liver
- Jaundice at (or shortly after) birth
- Learning difficulties and intellectual decline
- Seizures
- Slurred, irregular speech
- Sudden loss of muscle tone that may lead to falls
- Tremors
- Trouble moving the eyes up and down




Exams and Tests
A blood or bone marrow test can be done to diagnose types A and B. The test can tell who has the disease, but does not show if you are a carrier. DNA tests can be done to diagnose carriers of types A and B.
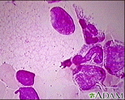
A skin biopsy is usually done to diagnose types C and C1. A technician watches how the skin cells grow, move, and store cholesterol. DNA tests may also be done to look for the 2 genes that cause this type of the disease.

Other tests might include:
- Bone marrow aspiration
- Liver biopsy (usually not needed)
- Slit-lamp eye exam
- Tests to check level of ASM

Treatment
At this time, there is no effective treatment for the neurologic symptoms of type A. Enzyme replacement with olipudase alfa was approved by the FDA in 2022 to treat non-central nervous system symptoms.
Bone marrow transplants may be tried for type B. Enzyme replacement with olipudase alfa is approved by the FDA to treat non-central nervous system symptoms of type B. Researchers continue to study additional possible treatments, such as gene therapy.
A medicine called miglustat is available for the nervous system symptoms of type C.
High cholesterol may be managed with a healthy, low-cholesterol diet or medicines. However, research does not show that these methods stop the disease from getting worse or change how cells break down cholesterol. Medicines are available to regulate or relieve many symptoms, such as sudden loss of muscle tone and seizures.
Support Groups
These organizations can provide support and more information on Niemann-Pick disease:
- National Institute of Neurological Disorders and Stroke -- www.ninds.nih.gov/health-information/disorders/lipid-storage-diseases
- National Niemann-Pick Disease Foundation -- nnpdf.org
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/niemann-pick-disease-type-c/
Outlook (Prognosis)
NPD type A is a severe disease. It usually leads to death by age 2 or 3.
Those with type B may live into late childhood or adulthood.
A child who shows signs of type C before age 1 may not live to school age. Those who show symptoms after entering school may live into their mid-to-late teens. Some may live into their 20s.
When to Contact a Medical Professional
Make an appointment with your health care provider if you have a family history of Niemann-Pick disease and you plan to have children. Genetic counseling and screening is recommended.
Contact your provider if your child has symptoms of this disease, including:
- Developmental problems
- Feeding problems
- Poor weight gain
Prevention
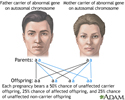
All types of Niemann-Pick are autosomal recessive. This means that both parents are carriers. Each parent has one copy of the variant gene without having any signs of the disease themselves.

When both parents are carriers, there is a 25% chance that their child will have the disease and a 50% chance that their child will be a carrier.
Carrier detection testing is only possible if the genetic defect is identified. The defects involved in types A and B have been well-studied. DNA tests for these forms of Niemann-Pick are available.
Genetic defects have been identified in the DNA of many people with type C. It may be possible to diagnose people who carry the abnormal gene.
A few centers offer tests to diagnose a baby still in the womb.
References
Kliegman RM, St. Geme JW, Blum NJ, et al. Defects in metabolism of lipids. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 106.
Turnpenny PD, Ellard S, Cleaver R. Inborn errors of metabolism. In: Turnpenny PD, Ellard S, Cleaver R, eds. Emery's Elements of Medical Genetics and Genomics. 16th ed. Philadelphia, PA: Elsevier; 2022:chap 18.
Review Date: 11/6/2024
Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 All rights reserved.
All rights reserved.