

Homocystinuria
Cystathionine beta-synthase deficiency; CBS deficiency; HCY
Homocystinuria is a genetic disorder that affects the metabolism of the amino acid methionine. Amino acids are the building blocks of all proteins in the body.

Causes
Homocystinuria is inherited in families as an autosomal recessive trait. This means that the child must inherit a non-working copy of the gene from each parent to be seriously affected.

People with homocystinuria have several physical features in common with Marfan syndrome, including skeletal and eye changes, but they are different conditions.
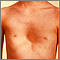
Symptoms
Newborn infants appear healthy. Early symptoms, if present, are not obvious.
Symptoms may occur as mildly delayed development or faltering weight. Increasing visual problems may lead to diagnosis of this condition.

Other symptoms include:
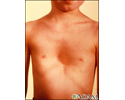
- Chest deformities (pectus carinatum, pectus excavatum)
- Flush across the cheeks
- High arches of the feet
- Intellectual disability
- Knock knees
- Long limbs
- Mental disorders
- Nearsightedness
- Spidery fingers (arachnodactyly)
- Tall, thin build

Exams and Tests
In all states, children are screened for homocystinuria as part of newborn testing.
Your health care provider may notice that your child is tall and thin.
Other signs include:
- Curved spine (scoliosis)
- Deformity of the chest
- Dislocated lens of the eye

If there is poor or double vision, an eye doctor (ophthalmologist) will perform a dilated eye exam to look for dislocation of the lens or nearsightedness.
There may be a history of blood clots. Intellectual disability or mental illness is also possible.
Tests that may be ordered include any of the following:
- Amino acid screen of blood and urine
- Genetic testing
- Homocysteine level
- Liver biopsy and enzyme assay
- Skeletal x-ray
- Skin biopsy with a fibroblast culture
- Standard ophthalmic exam



Treatment
There is no cure for homocystinuria. About half of people with the disease respond to vitamin B6 (also known as pyridoxine).

Those who do respond will need to take vitamin B6, B9 (folate), and B12 supplements for the rest of their lives. Those who do not respond to supplements will need to eat a low-methionine diet. Most will need to be treated with trimethylglycine (a medicine also known as betaine).
Neither a low-methionine diet nor medicine will improve existing intellectual disability. Medicine and diet should be closely monitored by a provider who has experience treating homocystinuria.
Support Groups
More information and support for people with homocystinuria and their families can be found at:
- National Organization for Rare Disorders -- rarediseases.org/rare-diseases/homocystinuria-due-to-cystathionine-beta-synthase-deficiency/
- HCU Network America -- hcunetworkamerica.org
Outlook (Prognosis)
Although no cure exists for homocystinuria, vitamin B therapy can help about half of people affected by the condition.
If the diagnosis is made in childhood, starting a low-methionine diet quickly may prevent some intellectual disability and other complications of the disease. For this reason, all states screen for homocystinuria in all newborns.
People whose blood homocysteine levels continue to rise are at increased risk for blood clots. Clots can cause serious medical problems and shorten lifespan.
Possible Complications
Most serious complications occur due to blood clots. These episodes can be life threatening.
Dislocated lenses of the eyes can seriously damage vision. Lens replacement surgery may be needed.
Intellectual disability is a serious outcome of the disease. But, it can be reduced if diagnosed early.
When to Contact a Medical Professional
Contact your provider if you or a family member shows symptoms of this disorder, especially if you have a family history of homocystinuria.
Prevention
Genetic counseling is recommended for people with a family history of homocystinuria who want to have children. Prenatal diagnosis of homocystinuria is available. This involves culturing amniotic cells or chorionic villi to test for cystathionine synthase (the enzyme that is missing in homocystinuria).

If there are known gene conditions in the parents or family, samples from chorionic villus sampling or amniocentesis can be used to test for these defects before a baby is born.

References
Kliegman RM, St. Geme JW, Blum NJ, et al. Defects in metabolism of amino acids. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 105.
Schiff M, Blom HJ. Homocystinuria and hyperhomocysteinemia. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 193.
Review Date: 10/27/2025
Reviewed By: Anna C. Edens Hurst, MD, MS, Associate Professor in Medical Genetics, The University of Alabama at Birmingham, Birmingham, AL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team. Editorial update 04/14/2026.
© 1997- A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited.
 All rights reserved.
All rights reserved.