

Health Library
Sweat electrolytes test
Sweat test; Sweat chloride; Iontophoretic sweat test; CF - sweat test; Cystic fibrosis - sweat test
Sweat electrolytes is a test that measures the level of chloride in sweat. The sweat chloride test is the standard test used to diagnose cystic fibrosis (CF).
Images
I Would Like to Learn About:
How the Test is Performed
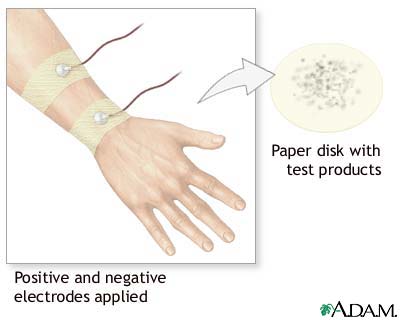
A colorless, odorless chemical that causes sweating is applied to a small area on an arm or leg. An electrode is then attached to the spot. A weak electrical current is sent to the area to stimulate sweating.
People may feel a tingling in the area, or a feeling of warmth. This part of the procedure lasts for about 5 minutes.
Next, the stimulated area is cleaned and the sweat is collected on a piece of filter paper or gauze, or in a plastic coil.
After 30 minutes, the collected sweat is sent to a hospital lab to be tested. The collection takes about 1 hour.
How to Prepare for the Test
No special steps are needed before this test.
How the Test will Feel
The test is not painful. Some people have a tingling feeling at the site of the electrode. This feeling may cause discomfort in small children.
Why the Test is Performed
Sweat testing is the standard method for diagnosing cystic fibrosis. People with cystic fibrosis have higher amounts of sodium and chloride in their sweat that are detected by the test.
Some people are tested because of symptoms they are having. In the United States, newborn screening programs test for cystic fibrosis. The sweat test is used to confirm these results.
Normal Results
Normal results include:
- A sweat chloride test result of less than 30 mmol/L in all populations means cystic fibrosis is less likely.
- A result between 30 to 59 mmol/L does not give a clear diagnosis. Further testing is needed.
- If the result is 60 mmol/L or greater, cystic fibrosis is present.
Note: mmol/L = millimole per liter
Normal value ranges may vary slightly among different labs. Talk to your health care provider about the meaning of your specific test results.
Some conditions, such as dehydration or swelling (edema) can affect the test results.
What Abnormal Results Mean
An abnormal test may mean that the child has cystic fibrosis. Results can also be confirmed by CF gene mutation panel testing.
Related Information
Cystic fibrosisReferences
Egan ME, Schechter MS, Voynow JA. Cystic fibrosis. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 454.
Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the Cystic Fibrosis Foundation. J Pediatr. 2017;181S:S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Siddiqi HA, Rabinowitz S, Axiotis CA. Laboratory diagnosis of gastrointestinal and pancreatic disorders. In: McPherson RA, Pincus MR, eds. Henry's Clinical Diagnosis and Management by Laboratory Methods. 24th ed. Philadelphia, PA: Elsevier; 2022:chap 23.
BACK TO TOPReview Date: 4/5/2025
Reviewed By: Charles I. Schwartz, MD, FAAP, Clinical Assistant Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, General Pediatrician at PennCare for Kids, Phoenixville, PA. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 | A.D.A.M., Inc. is accredited by URAC, for Health Content Provider (www.urac.org). URAC's accreditation program is an independent audit to verify that A.D.A.M. follows rigorous standards of quality and accountability. A.D.A.M. is among the first to achieve this important distinction for online health information and services. Learn more about A.D.A.M.'s editorial policy, editorial process and privacy policy. A.D.A.M. is also a founding member of Hi-Ethics. This site complies with the HONcode standard for trustworthy health information: verify here. |
The information provided herein should not be used during any medical emergency or for the diagnosis or treatment of any medical condition. A licensed medical professional should be consulted for diagnosis and treatment of any and all medical conditions. Links to other sites are provided for information only -- they do not constitute endorsements of those other sites. No warranty of any kind, either expressed or implied, is made as to the accuracy, reliability, timeliness, or correctness of any translations made by a third-party service of the information provided herein into any other language.
© 1997-
2026 A.D.A.M., a business unit of Ebix, Inc. Any duplication or distribution of the information contained herein is strictly prohibited.
All content on this site including text, images, graphics, audio, video, data, metadata, and compilations is protected by copyright and other intellectual property laws. You may view the content for personal, noncommercial use. Any other use requires prior written consent from Ebix. You may not copy, reproduce, distribute, transmit, display, publish, reverse-engineer, adapt, modify, store beyond ordinary browser caching, index, mine, scrape, or create derivative works from this content. You may not use automated tools to access or extract content, including to create embeddings, vectors, datasets, or indexes for retrieval systems. Use of any content for training, fine-tuning, calibrating, testing, evaluating, or improving AI systems of any kind is prohibited without express written consent. This includes large language models, machine learning models, neural networks, generative systems, retrieval-augmented systems, and any software that ingests content to produce outputs. Any unauthorized use of the content including AI-related use is a violation of our rights and may result in legal action, damages, and statutory penalties to the fullest extent permitted by law. Ebix reserves the right to enforce its rights through legal, technological, and contractual measures.
![]()